
α-アルミナ (α-Al2O3) の亀裂破壊のシミュレーション
有限温度にある物質中の原子のダイナミクス、電子状態などを理論的手法、主として計算機シミュレーションの手法を用いて調べています。 また、新しいシミュレーション手法の開発も精力的に行っています。
第一原理シミュレーションによる物質科学
半導体物質の亀裂破壊と応力腐食、化合物半導体中の圧力誘起構造相転移、ペロフスカイト型酸化物中のプロトン伝導機構、高圧化における液体・アモルファス物質の構造と電子状態、液体カルコゲン混合系の非金属・金属転移、固体電解質中のイオン拡散機構など高温状態にある物質中の種々の現象を理論的・計算機シミュレーション的手法を用いて調べています。
シミュレーション新手法の開発と高速化
オーダーN電子状態計算法の開発、第一原理・古典ハイブリッド分子動力学法の開発、並列化分子動力学法の開発、第一原理電子状態計算の並列化、グラフィックス・アニメーションによる可視化等に関する研究開発を行っています。
α-アルミナ (α-Al2O3) の亀裂破壊のシミュレーション
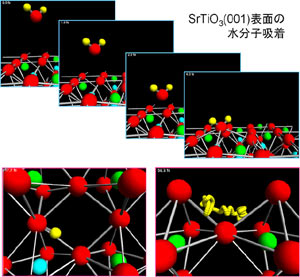
ペロブスカイト型酸化物中のH+拡散に関するシミュレーション
SrTiO3中のプロトンの安定位置 (左図) とプロトンの軌跡 (右図)
【関連分野・関連項目】
本研究テーマは、材料科学、物性物理学、固体物理学、有機・無機化学、計算化学などの分野と密接な関連があります。また、関連する (技術) 項目として、構造不規則系 (液体・アモルファス物質) の物性、固体酸化物型燃料電池、電極技術、水素吸蔵・水素エネルギー技術、半導体・デバイス技術、表面・界面物性などが挙げられます。
【共同研究、提供可能技術項目】
上記関連項目に関する実験的研究を行っている研究グループとの共同研究を希望します。 提供可能な技術項目として、第一原理分子動力学法による計算機シミュレーション関連技術、コンピューターグラフィックス技術などが挙げられます。